coMD is a methodology to generate all atomic transition pathways between any two structures/substates of a protein (described in MG9). The coMD script utilizes SHell , VMD , NAMD and MATLAB .
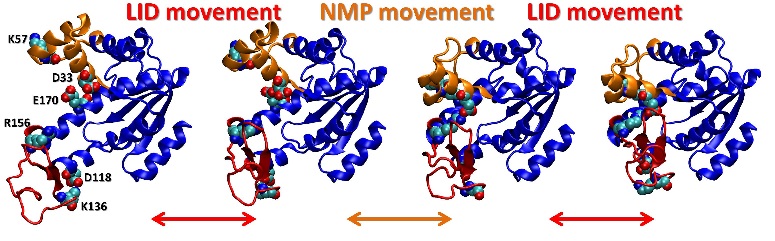
Since the aim is to simulate large systems we suggest to install all of these programs on a cluster/supercomputer. Please refer to the the schematic figure (Figure 2 in the Paper) below for a visual description where each program is required.
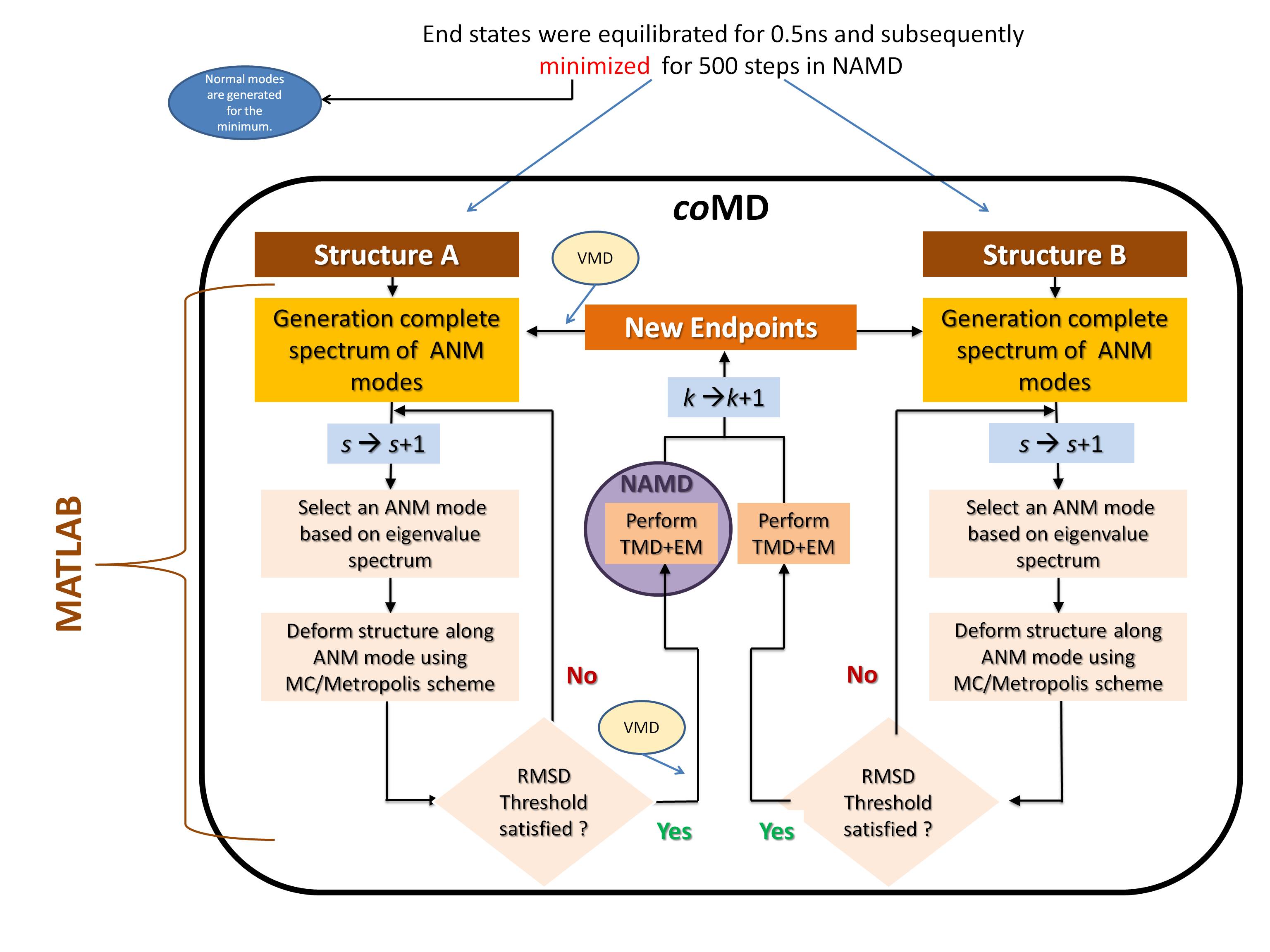
Figure 1: Schematic Description of coMD
The programs that are used to excecute each part are shown in the figure
If you benefited from coMD in your research, please cite the following paper: