

The Gur Lab has unveiled a fast, practical way to read molecular “movies” and pinpoint the forces that make proteins stick together or push apart.
The study, led by Associate Professor Mert Gur and conducted with postdoctoral scholar Mert Golcuk, was published in the Journal of Chemical Information and Modeling. It introduces a covariance-based workflow that detects both attractive and repulsive interactions across large protein interfaces with high efficiency.
“We built a tool that doesn’t just count close contacts; it captures the rhythm of motion between molecules,” Gur said. “If two residues move together, that’s a handshake; if they move in opposite directions, that’s a shove. Reading those patterns lets us zero in on what truly stabilizes or destabilizes protein complexes.”
Modern molecular dynamics (MD) simulations track tens of thousands to millions of atoms over hundreds of nanoseconds or longer. Traditional analyses often scan every possible residue pair with strict distance and angle cutoffs to find hydrogen bonds or salt bridges. This process is slow and misses destabilizing, like-charge repulsions that can be just as important for drug design and protein engineering.
The Gur Lab’s method brings a new lens to how researchers study molecular interactions. First, it builds an inter-protein covariance matrix — a map of how residues on different proteins move together or apart during a simulation. Second, it uses a distance filter to keep only residue pairs that actually come into contact. Third, it classifies what kind of interaction each pair forms — salt bridge, hydrogen bond, hydrophobic contact or repulsion — and how often it occurs.
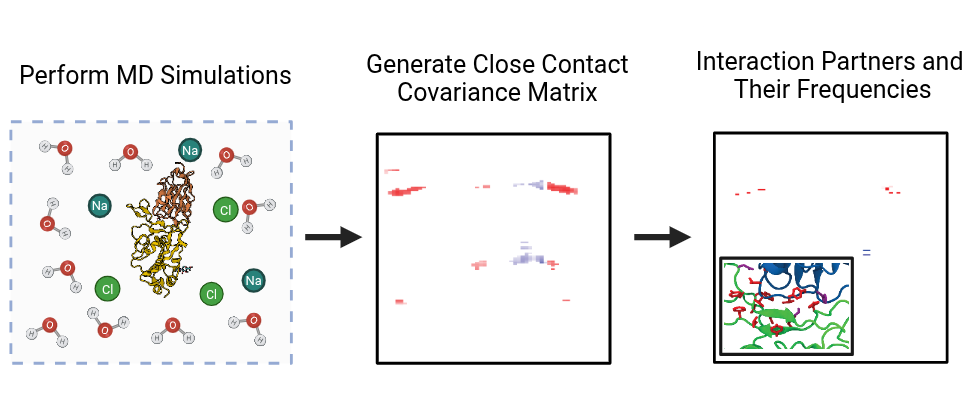
To demonstrate the approach, the team analyzed simulations of a SARS-CoV-2 nanobody (H11-H4) bound to spike proteins from multiple variants. The method recovered all major stabilizing contacts found by traditional, more labor-intensive scans. It also flagged new anticorrelated (repulsive) pairs that emerged in the Beta and Omicron variants.
Beyond accuracy, the big win is speed and scalability. By filtering for correlated, close contacts first, the team cut the number of residue-pair calculations by an order of magnitude. “When simulations get big and long, analysis can become a bottleneck,” Gur said. “Our approach focuses computing power — and human attention — on the parts of the interface that matter most.”
Designed to be user-friendly and extensible, the workflow can be applied to antibody–antigen systems, molecular motors on microtubules, and large multiprotein assemblies. The authors have made their code and data openly available to the biological community.
“We hope this invites students and collaborators from many backgrounds,” Gur added. “If you’re excited by big data, physics, and biology, this is a powerful entry point into computational discovery.”